| Issue |
Natl Sci Open
Volume 5, Number 3, 2026
|
|
|---|---|---|
| Article Number | 20260013 | |
| Number of page(s) | 15 | |
| Section | Materials Science | |
| DOI | https://doi.org/10.1360/nso/20260013 | |
| Published online | 03 April 2026 | |
RESEARCH ARTICLE
In-situ organic cation evolution driven sequential crystal transformation in hybrid metal halides
School of Materials Science and Engineering, Tianjin Key Laboratory of Metal and Molecular Materials Chemistry, Frontiers Science Center for New Organic Matter, Academy for Advanced Interdisciplinary Studies, Nankai University, Tianjin 300350, China
* Corresponding author (email: This email address is being protected from spambots. You need JavaScript enabled to view it.
)
Received:
26
January
2026
Revised:
27
February
2026
Accepted:
7
March
2026
Abstract
The properties of organic-inorganic hybrid metal halides (OIHMHs) strongly hinge on their crystal structures. However, structural diversity regulation based on organic-cation design or external stimuli is hindered by the limited diversity of available cations and the random structural changes. Here, we utilize in-situ sequentially generated organic cations from a stepwise reaction between dimethyl sulfoxide (DMSO) and acetone to drive a continuous and unidirectional single-crystal-to-single-crystal transformation in OIHMHs, enabling well-defined and predictable structural evolution. The intermediate dimethyl(2-oxopropyl)sulfonium and product trimethylsulfonium cations are sequentially generated and incorporated into the crystal lattice, giving rise to three types of structurally correlated OIHMHs in both Bi- and Sb-based systems. The resulting OIHMH materials preserve the inorganic photoactive unit while exhibiting systematic changes in their physicochemical properties, revealing a dynamic cooperative mechanism between evolving organic cations and the adaptive inorganic octahedral framework. This work demonstrates the feasibility of intermediate cations generated by organic reactions to serve as an underutilized A-site organic cation resource. Such an organic reaction-driven and inorganic framework-mediated strategy establishes a dynamic paradigm for structural evolution, opening a promising avenue toward precise structural modulations and diversified functionalities of OIHMH materials.
Key words: organic-inorganic hybrid metal halide / dissolution-reprecipitation transformation / dynamic structural reconstruction / in-situ cation evolution / adaptive inorganic framework
© The Author(s) 2026. Published by Science Press and EDP Sciences.
 This is an Open Access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
INTRODUCTION
Organic-inorganic hybrid metal halides (OIHMHs) are crystalline materials assembled through weak interactions between organic components and inorganic metal halide polyhedral frameworks [1–3]. OIHMHs exhibit exceptional optoelectronic properties [4,5], including high absorption coefficients [6,7], long carrier diffusion lengths [8–10], high photoluminescence quantum yields [11–13], and tunable bandgaps [14–16], which promise their broad application prospects in optoelectronic devices such as solar cells [17–19], light-emitting diodes [20–22], photodetectors [23,24] and lasers [25,26]. Fundamentally, the properties and functionalities of OIHMHs are governed by the structures and arrangements of both organic and inorganic components in their crystalline lattices [1,27,28]. Subtle variations in the size, geometry, and polarity of the organic components can deterministically tailor structural symmetry [29,30], lattice stability [31], and phase evolution [32]. Meanwhile, the dimensionality and distortion of the inorganic polyhedra directly determine band dispersion [16,33], charge mobility [34], and optical response [35]. Therefore, systematic development and fine control of OIHMH crystal structures are central to the design of high-performance functional materials and emerging optoelectronic devices. Current structural regulation of OIHMHs primarily relies on two types of strategies, one focused on component engineering within the internal lattice [31,36,37], and the other based on leveraging stimuli-responsive external conditions [2,38–40]. Both approaches aid in crystal reconstruction or phase transitions, yet each faces inherent limitations. Component engineering, exemplified by ion exchange, ligand modification, and solvent engineering, is gradually approaching saturation due to limited precursor variety. Furthermore, significant structural changes in the A-site or B-site cation components often result in poor correlation before and after evolution, making precise and targeted structural control difficult to achieve. Conversely, external stimuli (e.g., heat, pressure) typically lack selectivity and the structural changes are highly random [41]. Developing a strategy capable of in-situ expanding organic cation structures while enabling continuous, predictable, and strongly correlated crystal reconstruction is critically important for advancing structural diversification and the targeted construction of functional hybrid metal halide crystals.
Inherent dynamics within the OIHMH system confer significant migration and rearrangement capabilities of both organic and inorganic components, allowing for structural evolution that is not solely dictated by thermodynamic stability [42]. The synergistic regulation between the organic and inorganic components endows these hybrid systems with the essential characteristics of a dynamic chemical system [43], enabling continuous structural evolution in response to external conditions such as the solvent environment, temperature, or pressure. It has been recently reported that organic chemical reactions can proceed within the OIHMH mother liquors for the in-situ generation of A-site organic cations and successful incorporation into the OIHMH lattices [35,44–46], demonstrating the cooperation of the organic reactions with framework formation and crystal growth. However, the reported synthetic strategies primarily focus on “one-step” routes that yield structurally fixed OIHMH crystals, largely neglecting the intrinsic dynamic nature of these hybrid systems. Given the inherently multi-step, temporally evolving, and directional nature of organic chemical reactions, the reaction trajectories necessarily involve the sequential generation, transformation, and disappearance of various cationic species. This dynamic process creates a naturally tunable and evolutionarily sequential library of A-site cations for OIHMH construction. Such a sequential cation evolution aligns well with the intrinsic dynamic characteristics of OIHMHs, which can undergo continuous dissolution-reprecipitation [47,48] accompanied by internal framework rearrangements. By strategically coupling the stepwise continuity of multi-step organic reactions with the dissolution-reprecipitation mechanism of OIHMHs, it becomes possible to guide the sequential incorporation of cations with varying geometric sizes, polarizations, and configurations into the lattice. This approach might enable precise structural construction, fine-tuning, and even continuous transformations in OIHMHs.
In this work, we take advantage of the stepwise condensation reaction system of dimethyl sulfoxide (DMSO) and acetone as the precursor solution for sequential construction of bismuth (Bi)- and antimony (Sb)-based OIHMHs. This approach establishes a tight coupling between OIHMH crystal growth and the in-situ generated and dynamically evolving organic cations. Specifically, the condensation reaction of DMSO and acetone generates the intermediate cation dimethyl(2-oxopropyl)sulfonium (DOPS+) and the product cation trimethylsulfonium (TMS+) [49] in sequence. The evolving concentration gradients and distinct coordination preferences of these cations direct their stepwise incorporation into the OIHMH lattice, providing the intrinsic driving force for crystal structural reorganization. This dynamic process enables continuous and precisely controllable single-crystal-to-single-crystal (SCSC) transformations of OIHMHs. Through comprehensive reaction trajectory analysis and monitoring, three sets of temporally sequential OIHMH crystals could be successfully captured: an initial OIHMH I containing only the intermediate cations DOPS+, an OIHMH II co-existing with both DOPS+ and TMS+, and the final OIHMH III exclusively composed of the end-product TMS+ as the organic cation (Scheme 1). Importantly, the successful capture of intermediate single crystals provided atomic-level structural insights into the reactive intermediate cations, simultaneously revealing their dual function as essential lattice-building units and as the intrinsic drivers of structural reorganization. Introducing dynamic covalent chemistry concepts to weakly-interacting OIHMH materials allows for the non-destructive and global reconstruction of the crystal structure under mild conditions. This strategy provides a highly promising ‘reaction-driven’ approach to the construction of functionally promising hybrid crystalline materials, leveraging the intrinsic evolution process of chemical reactions to manipulate solid-state structural transitions, and thus opening a new research paradigm for chemical kinetics-based crystal engineering.
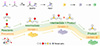 |
Scheme 1 Schematic illustration of the correspondence between the sequential generation of intermediate and product cations from the DMSO and acetone condensation reaction and the sequential formation of the three corresponding OIHMH crystals. |
RESULTS AND DISCUSSION
Synthesis and structural transformation
In the presence of hydrobromic (HBr) acid, the reaction between DMSO and acetone proceeds through a well-defined multistep condensation pathway, sequentially yielding DOPS+ intermediate and the TMS+ product [49,50]. These two organosulfur cations exhibit highly correlated structures and can each stably incorporate into the OIHMH lattice as A-site organic components [45]. Consequently, this stepwise condensation reaction provides a time-evolving pool of cation candidates, offering the possibility of continuous modulation of the cations incorporated during the OIHMH crystal formation. The intrinsic physicochemical characteristics of the reaction system further create conditions ideally suited for OIHMH crystallization. Owing to the significant density difference between DMSO (1.10 g/cm3) and acetone (0.79 g/cm3), stable stratified solvent layers could be formed, establishing a supersaturated interface that promotes orderly nucleation and growth of OIHMH crystals. Meanwhile, HBr acid dissolves preferentially in DMSO, and the resulting solution effectively dissolves diverse metal halide salts, thereby forming a robust precursor environment for OIHMHs [51]. In contrast, the resulting OIHMHs display relatively low solubility in acetone [52]. As acetone gradually diffuses into the DMSO and HBr layer, slow crystallization of OIHMHs is induced. These features together motivate the design of a liquid-phase diffusion crystallization system in a stratified configuration [53], with an upper acetone layer overlying a DMSO solution containing HBr and metal salts (Figure 1a). This setup simultaneously satisfies the thermodynamic requirements of condensation reaction and the kinetic demands of OIHMH crystallization. It allows for the efficient interaction between acetone and DMSO to initiate the organic reaction, while the subsequent slow diffusion process ensures continuous in-situ generation of candidate cations throughout crystal growth. After the system is left undisturbed at room temperature for several days, crystals with well-defined morphology gradually precipitate (see experimental section in Supporting Information). Single crystal X-ray diffraction (SCXRD) analysis identifies these crystals as three distinct types of OIHMHs, crystallized in the C2/c, Pnma and R-3m space groups, respectively (Figure 1b). Structural analysis clearly reveals that DOPS+ and TMS+ are successfully captured within their corresponding OIHMH lattices, directly verifying the in-situ synthesis and sequential incorporation of organic cations during crystal formation.
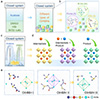 |
Figure 1 Multistep structural evolution of OIHMHs driven by the in-situ condensation reaction. (a) Schematic illustration for the formation of various OIHMH crystal structures achieved by direct employment of the DMSO and acetone reaction pathway as the liquid diffusion crystallization system. (b) Structures of three distinct types of obtained OIHMHs, crystallized in the C2/c, Pnma and R-3m space groups, respectively. (c) Schematic illustration for the purification of the three OIHMH crystal types achieved by designing the DMSO and acetone reaction system as a vapor diffusion crystallization. (d) The sequential incorporation of the intermediate and product cations into the A-sites constructs three distinct OIHMH structures. (e) The minimum asymmetric units and structural transformation sequence of the three obtained OIHMH types. H atoms are omitted for clarity. |
The liquid-phase diffusion crystallization system enables the simultaneous obtaining of three distinct OIHMH crystals, each incorporating the cations sequentially generated from the condensation reaction between DMSO and acetone. However, the concurrent observation of these three crystals in the same crystallization environment made it difficult to directly correlate each organic reaction stage with the formation of its corresponding OIHMH structure and thereby obscure the role of cationic generation sequence in directing crystal evolution. A vapor-phase evaporation crystallization system [54] has thus been constructed for further analysis of the intrinsic relationship between the sequence of cation generation and the formation of OIHMH crystals. This setup involves the placement of acetone in an outer container and the precursor of DMSO and HBr solution in an inner vial (Figure 1c). Vaporized acetone from the outer chamber slowly diffused into the inner solution containing the metal precursor, avoiding instantaneous bulk mixing of reactants. This approach significantly retards the rate of the condensation reaction, providing an expanded temporal window during which sequentially generated cations could interact selectively with the inorganic framework (Figure 1d). Given the pronounced temperature dependence of the reaction progression and rate [55,56], regulation of the crystallization temperature and reaction time allows for sequential participation of each cation in lattice construction according to its specific reaction order. By subjecting the crystalline system to low (15 °C), medium (30 °C), and high temperatures (60 °C), the reaction process is compartmentalized, enabling different cationic species to dominate the crystallization of OIHMHs within their respective thermal windows. SCXRD analysis confirms the formation of a unique OIHMH crystal within each temperature window. The crystal obtained at 15 °C is formulated as (DOPS)4[(Bi/Sb)Br6]Br (OIHMH I), incorporating only the intermediate DOPS+. At 30 °C, the as-formed crystal corresponds to (DOPS)2(TMS)[(Bi/Sb)Br6] (OIHMH II), which contains both DOPS+ and TMS+ cations. At 60 °C, the exclusive presence of product cation TMS+ has been identified, as (TMS)4[(Bi/Sb)Br6]Br (OIHMH III) (Figure 1e, Table S1). All three compounds crystallize in zero-dimensional (0D) configurations, but in very different space groups of the C2/c, Pnma, and R-3m, respectively, matching those obtained from the liquid-phase diffusion crystallization experiment. Powder X-ray diffraction (PXRD) patterns of these three OIHMH types agree well with the simulated patterns, showing the considerable phase purity (Figure S1).
Characterizations and physicochemical properties
Complete dissolution of the high-purity OIHMH crystals liberates their organic components into the solution, allowing for the precise recognition and validation of the organic cation structures present in each crystal. 1H nuclear magnetic resonance (1H NMR) spectra of the dissolved OIHMH samples confirm the presence and identity of the in-situ synthesized cationic species within their respective crystal lattices. Both cations of DOPS+ (in OIHMH I) and the TMS+ (in OIHMH III) serve as the sole organic component in the corresponding OIHMHs. The chemical shifts and integrated peak areas are consistent with the expected hydrogen environments within the organic components of each sample (Figure 2a, Figure S2a, c). The crystal lattice of OIHMH II contains both DOPS+ and TMS+, present in a 2:1 molar ratio within its asymmetric unit. Consistent with this stoichiometry, its solution-state 1H NMR spectra exhibit proton signal integrations that reflect the same ratio. Although the S-CH3 resonances of DOPS+ and TMS+ appear in close proximity and display slight coupling, their signals remain sufficiently resolved to permit reliable integration (Figure 2a and Figure S2b). The resulting integration aligns with the crystallographic composition, confirming the simultaneous incorporation of both cations within the lattice. Furthermore, 13C NMR spectrum of OIHMH II exhibited the characteristic carbonyl (C=O) signal of DOPS+. All carbon atoms in distinct chemical environments give rise to well-resolved and assignable resonance peaks (Figure 2b), further demonstrating the stable incorporation of both DOPS+ and TMS+ organic cations into the lattice structure of OIHMH II. Clear spectral characteristics corresponding to the DOPS+ and TMS+ have also been observed in the mass spectra (MS) of the three dissolved OIHMH samples (Table S2), also confirming the organic component composition within their crystal lattices.
 |
Figure 2 Spectroscopic characterizations. (a) 1H NMR spectra of Sb-based OIHMH I, OIHMH II and OIHMH III, in DMSO-d6. (b) 13C NMR spectra of the Bi/Sb-based OIHMH II dissolved samples, in DMSO-d6. (c) FTIR spectra of the three Sb-based OIHMH powders. (d) UV-vis transmission spectra and Eg comparison of the three types of Bi/Sb-based OIHMHs. (e) Comparison of the thermal decomposition temperature of the three types of Bi/Sb-based OIHMHs. |
Fourier transform infrared spectroscopy (FTIR) of the three OIHMHs further corroborates this correspondence, as the characteristic absorption peaks precisely match the organic components contained within the crystal structures (Figure 2c and Figure S3). A characteristic peak corresponding to the C–S single bond stretching vibration is consistently observed around 1040 cm−1 in all samples [57], indicating the universal incorporation of sulfonium cations within the crystal lattices. The sharp peaks detected at ~1700 cm−1 in the spectra of OIHMH I and OIHMH II are attributed to the carbonyl (C=O) group in the DOPS+ [58]. In contrast to OIHMH I and OIHMH II, the FTIR spectra of OIHMH III do not exhibit the –CH2– stretching vibration peak (at approximately 2850 cm−1). This absence is attributed to the exclusive presence of the TMS+ within the OIHMH III lattice. These spectral features clearly delineate the distinct structural characteristics of the A-site organic components of all three crystal types. Furthermore, it has been demonstrated that the two classes of sulfonium cations, generated in situ in sequence, successively participate in the lattice construction. Comparative analysis of the A-site cation components across the three OIHMH types establishes a direct correlation between the organic reaction stages and the formation of OIHMH crystals: DOPS+ governs the formation of OIHMH I, the coexistence of DOPS+ and TMS+ leads to OIHMH II, and the eventual dominance of TMS+ drives the emergence of OIHMH III (Scheme 1).
The structural variations among the three OIHMHs, directly driven by the differences in their organic components, give rise to systematic distinctions in their physicochemical properties. Within each metal system, the ultraviolet-visible (UV-vis) transmission spectra of the three OIHMHs exhibit highly similar absorption features, including both spectral profiles and peak positions (Figure 2d). In contrast, pronounced differences could be noticed between Bi-based and Sb-based OIHMHs. This comparison indicates the primary dictation of optical absorption by the inorganic polyhedra, which impose consistent electronic transition characteristics within the same metal species. Direct optical band gaps (Eg) extracted via Tauc analysis [59,60] further support this conclusion (Figure S4, Table S3). Sb-based OIHMHs (Eg(Sb (I)) = 3.03 eV, Eg(Sb (II)) = 3.01 eV, Eg(Sb (III)) = 2.98 eV) consistently display larger band gaps than their Bi-based counterparts (Eg(Bi (I)) = 3.00 eV, Eg(Bi (II)) = 2.97 eV, Eg(Bi (III)) = 2.96 eV). This trend arises from the lower energy of the Sb3+ 5p orbitals compared to that of Bi3+ 6p orbitals, which elevates the conduction band minimum in Sb-based OIHMHs and consequently widens the band gaps [61,62]. Notably, a monotonic band gap reduction is observed across the three sequentially constructed OIHMH crystals within each metal system, demonstrating that the progressive conversion of organic cations can systematically modulate the metal-halide bonding environment and the organization of the inorganic framework. This effect shifts the band-edge positions without altering the fundamental electronic transition mechanisms, which allows for the effective tuning of the structural bandgap.
Thermogravimetric analysis (TGA) reveals a progressive enhancement in the thermal stability of the three types of OIHMHs (Figure 2e and Figure S5, Table S4). The OIHMH I containing intermediate DOPS+ undergoes structural collapse at approximately 110 °C (Sb (I)) and 142 °C (Bi (I)). In contrast, OIHMH II, which incorporates DOPS+ and product TMS+, exhibits greater stability, resisting decomposition until around 164 °C (Sb (II)) and 168 °C (Bi (II)). The OIHMH III with the final product TMS+ demonstrates the highest thermal stability, with its thermal decomposition initiating at about 207 °C (Sb (III)) and 187 °C (Bi (III)). The progressive increase in thermal stability is directly attributable to the compositional and structural evolution of the organic components within these OIHMHs [63]. The evolution involves the gradual conversion of less stable intermediate cations into the most stable final product cations. Concurrently, the inorganic components adapt to these organic transformations through complementary adjustments in lattice organization, involving both expansion and contraction [64]. A comparative analysis of the two metal systems reveals distinct sensitivities to cation evolution. The maximum band gap difference among the three Bi-based OIHMHs is approximately 0.04 eV, while the Sb-based OIHMHs display a larger variation of about 0.05 eV. Similarly, the maximum difference value in thermal decomposition temperature increases from 45 °C (for the Bi-based OIHMHs) to ~100 °C (for the Sb-based OIHMHs). These comparisons indicate that, within the DMSO and acetone reaction system driving sequential cation evolution, the progressive incorporation of these cations exerts a more pronounced impact on thermal stability than light absorption. Moreover, the systematic structural evolution leads to more distinct and coherent changes in the physical properties of the Sb-based OIHMHs compared to their Bi-based counterparts.
Transformation tracking and mechanism exploration
The three Sb-based OIHMH crystals exhibit yellow photoluminescence (PL) under UV excitation, with broad emission bands and large Stokes shifts in their spectra (Figure 3a and Figure S6, Table S5). The corresponding time-resolved PL decay curves are well-fitted by the mono-exponential function, with microsecond-scale lifetimes (Figures 3b and S7, Table S5). These combined spectral characteristics suggest that the PL of these three Sb-based OIHMHs originates from self-trapped exciton (STE) emission [52], which is strongly correlated with the structural organization of the inorganic components within OIHMHs [65,66]. The variations in the spacing between adjacent inorganic octahedral units and the extent of Sb-Br octahedral distortion among these three Sb-based OIHMH crystals lead to differences in their maximum emission wavelengths and PL quantum yields (PLQY) [67] (Figure 3a, b and Figure S8, Table S5). Among them, OIHMH I (Sb (I)) exhibits an optimal emission peak at ~563 nm, with a PLQY up to 100% at its optimal excitation wavelength. OIHMH II (Sb (II)) shows a maximum emission peak at ~566 nm with a PLQY of 26.6%, while the final product OIHMH III (Sb (III)), displays an emission maximum at ~560 nm and achieves a PLQY of 41.2%. Upon excitation with a femtosecond laser (Figure S9) [68] at wavelengths ranging from 720 to 800 nm, two-photon fluorescence (TPF) signals of these three types of OIHMH single crystals could be recorded (Figure S10). The TPF spectra of these three OIHMHs provide further confirmation of the variations in both their maximum emission wavelength and luminescence intensity across different crystals (Figure 3c) [69]. The variation of TPF intensity with incident polarization angle differs significantly among the three crystals, which can be attributed to their distinct crystallographic symmetries (Figure S11) [68]. The TPF intensity scales quadratically with the input laser power, confirming the two-photon characteristics of the TPF emission (Figure S12).
 |
Figure 3 Variation of optical properties during the crystal transition. (a) Normalized PL spectra of the three Sb-based OIHMH crystals. (b) Comparison of the PL decays and PLQYs. (c) Comparison of TPF, at their optimal incident laser wavelength, with a laser power of 50 mW. (d) Crystal transition of the Sb-based OIHMH monitored through UV-light crystal images and corresponding PXRD patterns. |
Leveraging the distinctive luminescence of Sb-based OIHMHs, we directly track the crystal structural reconstruction driven by the progressing organic reaction through monitoring PL changes during vapor-phase evaporation. Time-resolved optical and morphological observations, corroborated by powder XRD analysis, elucidate a sequential structural evolution mechanism in OIHMHs. This mechanism is triggered by sulfonium cations that are progressively generated from the reaction of DMSO and acetone (Figure 3d). At the onset of the reaction, the initially yielded intermediate cation DOPS+ spontaneously assembles with metal halides in the system to form OIHMH I, which crystallizes first from solution and exhibits bright yellow PL. As the reaction proceeds, DOPS+ is gradually consumed while the product cation TMS+ is continuously produced. When the concentration of TMS+ becomes sufficient to co-interact with DOPS+ and the inorganic polyhedra, OIHMH II with weaker orange PL begins to nucleate and grows epitaxially on the surface of OIHMH I. Powder XRD analysis at this stage reveals coexisting characteristic reflections from both OIHMH I and OIHMH II crystals, confirming the initiation of the crystal reconstruction process. As more DOPS+ in the solution are converted into the final product TMS+, the concentration of DOPS+ further decreases, leading to the gradual and complete dissolution of OIHMH I. The system then enters a growth stage dominated by OIHMH II. Subsequently, the continuously rising concentration of TMS+ triggers a new round of selective nucleation. Smaller OIHMH III crystals with hexagonal platelet morphology appear on the surface of OIHMH II and progressively assemble into stable polycrystalline polyhedra. Eventually, OIHMH II fully dissolves, leaving only pure OIHMH III in the system, marking the completion of the structural transformation.
Throughout the crystal transformation process, the organic cations generated at each reaction stage selectively couple with the lattice framework following the reaction sequence. The stepwise generation, accumulation, and consumption of these distinct cations dictate both the temporal window and the directionality of the structural transition. Meanwhile, the inorganic [SbBr6]3−/[BiBr6]3− octahedra are dynamically recycled during the structural transition [51], participating in the dissolution of the initial crystal and the construction of the subsequent one, thereby enabling continuous structural evolution. From a microscopic perspective, the driving force of the OIHMH phase transition primarily originates from the stepwise reactions of the organic cations. Once the intermediate cations embedded within the crystal undergo subsequent reactions, local lattice collapse occurs, triggering the dissolution of the parent crystal and generating surface vacancies and structural defects. The immediate vicinity of these vacancies and defects is enriched with uncoordinated or weakly coordinated inorganic octahedra, providing high-energy sites and local precursor environments for the insertion of newly formed cations, thus becoming the preferred nucleation locations. As the new cations enter the lattice and form stable interactions, the inorganic framework reorganizes accordingly, promoting interface-mediated epitaxial growth and achieving a unidirectional and continuous transition. Therefore, the strong electrostatic interactions provided by the metal halide polyhedra endow the lattice with the capacity to stably capture various organic cations, while the sequential formation of organic sulfonium cations determines the structural evolution pathway of OIHMHs. The synergistic interplay of these features governs a dynamic dissolution-reassembly-recrystallization process, enabling multistage, controllable structural reconfiguration in OIHMHs while preserving the chemical identity of inorganic building blocks.
Fundamentally, the reconstruction of OIHMHs driven by in-situ cation evolution is governed by the intrinsic balance between flexibility and robustness of the inorganic framework. This adaptive yet stable lattice enables the effective incorporation, exchange, and subsequent release of diverse organic cations. These inorganic octahedra accommodate evolving organic cations through adjustment in local distortions, stacking arrangements, and symmetry modes, thereby ensuring continuous structural compatibility throughout the transformation. The three types of OIHMHs exhibit a systematic increase in their packing arrangements and symmetry levels. The sequential evolution from monoclinic (C2/c) to orthorhombic (Pnma) packing, ultimately stabilizing in the trigonal (R-3m) crystal system, reflects the lattice’s adaptive response to the stepwise evolution of the organic cations. As the organic components in these structures evolve from isolated chain-like DOPS+ to coexisting DOPS+ and TMS+, and finally to exclusive presence of TMS+, the arrangement of [SbBr6]3−/[BiBr6]3− octahedra undergoes a corresponding structural transformation. The two dominant angular orientations observed in OIHMH I and OIHMH II (Figure 4a, b, Figures S13 and S14) eventually converged into a single and uniform octahedral orientation in OIHMH III (Figure 4c and Figure S15). Concurrently, the metal-metal distance between adjacent metal centers progressively decreases. This trend reflects the combined effects of the spatial volume, conformational freedom, and localization of the different organic cations within the crystal lattice. The presence of chain-like DOPS+ in OIHMH I drives a laterally compact packing of metal octahedra while maintaining a relatively large interlayer spacing (Figure 4a and Figure S16). Upon transformation to OIHMH II, the lattice incorporates both the highly disordered DOPS+ and the smaller TMS+ cations (Figure S17). This complex organization of organic components promotes greater flexibility in the arrangement of the inorganic octahedra, leading to a higher degree of freedom for rearrangement (Figure 4b and Figure S17). Ultimately, when the crystal structure stabilizes as OIHMH III, the lattice exclusively contains the TMS+. Given the high concentration of product cations and the comparatively limited number of metal octahedra in this crystal, these octahedra cooperatively interact with free halogen species within the system to stabilize the abundant product TMS+ cations. As a result, the adjacent metal octahedra exhibit the most uniform distribution, and the overall arrangement demonstrates greater structural regularity (Figure 4c and Figure S18).
 |
Figure 4 Structural comparison of the inorganic components among the three types of OIHMHs. The metal–metal distances between adjacent inorganic components of Sb-based OIHMH I (a), OIHMH II (b) and OIHMH III (c). The bond lengths, bond angles, and distortions of the [SbBr6]3− and [BiBr6]3− inorganic components across OIHMH I (d), OIHMH II (e), and OIHMH III (f). |
Through steric interactions, these structurally diverse organic cations further induce different degrees of distortion in the metal octahedra within the OIHMH lattice [31]. The level of distortion, as defined by different bond lengths and angles of the metal octahedra (Figure 4d–f), with 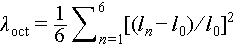 and
and 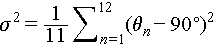 , where ln represents the length of each Sb–Br or Bi–Br bond, l0 is the average length of all Sb–Br or Bi–Br bonds, and θn is the angle formed by each Br–Sb–Br or Br–Bi–Br bond [70]. While [BiBr6]3− and [SbBr6]3− octahedra differ in absolute bond lengths due to their distinct metal ionic radii, the distortion degrees and evolutionary trends are highly consistent across the three types of crystals. Among them, OIHMH II exhibits the most pronounced metal octahedral distortion ([SbBr6]3−: λcot = 23.9 × 10−5 and σ2 = 3.59 deg2, [BiBr6]3−: λcot = 8.9 × 10−5 and σ2 = 3.73 deg2) (Figure 4e, Table S6), which could be attributed to local geometric perturbations caused by the size disparity between the two types of cations. OIHMH III possesses minimal distortion, with its metal octahedra closely approximating an ideal geometry, characterized by λcot = 0 and σ2 = 0.04 (in [SbBr6]3−) or 0.03 (in [BiBr6]3−) deg2 (Figure 4f, Table S6), owing to the sole presence of highly symmetric TMS+ in its lattice. The metal octahedra in OIHMH I show moderate distortion ([SbBr6]3−: λcot = 1.2 × 10−5 and σ2 = 1.84 deg2, [BiBr6]3−: λcot = 0.92 × 10−5 and σ2 = 2.16 deg2) (Figure 4d, Table S6). It is noteworthy that the exceptionally high PLQY observed in Sb-based OIHMH I is closely associated with a suitable degree of octahedral distortion, coupled with an optimal intermetal distance [71]. Moreover, octahedral distortions alter the distances between metal octahedra, thereby modulating local orbital overlap and orbital coupling strength, which enables precise tuning of the optical bandgap in OIHMHs. Overall, the in-situ generated organic cations govern the stepwise reconstruction of the inorganic framework through differences in their size, symmetry, and steric hindrance. Meanwhile, both distortion and reorganization of the inorganic framework serve to sterically and energetically stabilize the incorporation of diverse organic components. This organic-driven and inorganic-adaptive coupling mechanism provides a general principle for rationally designing hybrid materials with controllable structural evolution.
, where ln represents the length of each Sb–Br or Bi–Br bond, l0 is the average length of all Sb–Br or Bi–Br bonds, and θn is the angle formed by each Br–Sb–Br or Br–Bi–Br bond [70]. While [BiBr6]3− and [SbBr6]3− octahedra differ in absolute bond lengths due to their distinct metal ionic radii, the distortion degrees and evolutionary trends are highly consistent across the three types of crystals. Among them, OIHMH II exhibits the most pronounced metal octahedral distortion ([SbBr6]3−: λcot = 23.9 × 10−5 and σ2 = 3.59 deg2, [BiBr6]3−: λcot = 8.9 × 10−5 and σ2 = 3.73 deg2) (Figure 4e, Table S6), which could be attributed to local geometric perturbations caused by the size disparity between the two types of cations. OIHMH III possesses minimal distortion, with its metal octahedra closely approximating an ideal geometry, characterized by λcot = 0 and σ2 = 0.04 (in [SbBr6]3−) or 0.03 (in [BiBr6]3−) deg2 (Figure 4f, Table S6), owing to the sole presence of highly symmetric TMS+ in its lattice. The metal octahedra in OIHMH I show moderate distortion ([SbBr6]3−: λcot = 1.2 × 10−5 and σ2 = 1.84 deg2, [BiBr6]3−: λcot = 0.92 × 10−5 and σ2 = 2.16 deg2) (Figure 4d, Table S6). It is noteworthy that the exceptionally high PLQY observed in Sb-based OIHMH I is closely associated with a suitable degree of octahedral distortion, coupled with an optimal intermetal distance [71]. Moreover, octahedral distortions alter the distances between metal octahedra, thereby modulating local orbital overlap and orbital coupling strength, which enables precise tuning of the optical bandgap in OIHMHs. Overall, the in-situ generated organic cations govern the stepwise reconstruction of the inorganic framework through differences in their size, symmetry, and steric hindrance. Meanwhile, both distortion and reorganization of the inorganic framework serve to sterically and energetically stabilize the incorporation of diverse organic components. This organic-driven and inorganic-adaptive coupling mechanism provides a general principle for rationally designing hybrid materials with controllable structural evolution.
CONCLUSIONS
In summary, we have developed a multistep organic reaction strategy for driving continuous structural transformation in OIHMHs. In-situ sequentially synthesized organic cations from DMSO and acetone serve both as lattice building blocks and intrinsic drivers of structural reorganization. The adaptive inorganic [SbBr6]3−/[BiBr6]3− octahedral framework responds through rotation, distortion, and spacing adjustment, accommodating the geometric and electronic evolution of diverse A-site cations. This cooperative organic-inorganic interplay ensures the crystals undergo unidirectional, sequential, and controllable transformations while preserving macroscopic morphology and long-range order. Temperature-controlled vapor-phase evaporation crystallization directly links cation evolution to lattice reconstruction and allows the isolation of structurally related OIHMHs. As a result, key physical and chemical properties of these OIHMHs, including optoelectronic behavior and thermal stability, can be precisely tailored through structural modulation. This organic reaction-driven approach to continuous OIHMH structural transformations reveals a general principle in which organic reaction kinetics and inorganic framework adaptability act synergistically, offering a new paradigm for designing hybrid crystalline materials with controllable structural evolution under mild conditions.
METHODS
Detailed materials and methods are available in the supplementary information online.
Data availability
Crystallographic data have been deposited in the Cambridge Crystallographic Data Center (CCDC) with the following accession numbers, 2386943, 2514342-2514346, which contain the crystal structures of (DOPS)4[Bi/SbBr6]Br, (DOPS)2(TMS)[Bi/SbBr6], (TMS)4[Bi/SbBr6]Br, respectively. These data can be obtained free of charge from the CCDC at https://www.ccdc.cam.ac.uk/structures/.
Funding
This work was supported by the National Natural Science Foundation of China (22475106, 22372082, 22494631), the National Key Research and Development Program of China (2022YFA1204500, 2022YFA1204504), the Natural Science Foundation of Tianjin City (23JCJQJC00110), the TCL Science and Technology Innovation Fund, and the Fundamental Research Funds for the Central Universities.
Author contributions
J.X. initiated and directed the research. X.-H.B., D.W. and M.C. co-supervised the project. W.H. and J.X. conceived and designed the experiments. W.H. carried out most of the synthetic and characterization experiments. J.G., X.G., W.L., R.Z., P.C., L.X., Q.L. and Z.W. contributed to the synthesis and characterization. W.H. and J.X. wrote the manuscript with the input from all the coauthors. All authors discussed the results and commented on the manuscript.
Conflict of interest
The authors declare no conflict of interest.
Supplementary information
Supplementary file provided by the authors. Access Supplementary Material
References
- Lee JW, Tan S, Seok SI, et al. Rethinking the A cation in halide perovskites. Science 2022; 375: eabj1186. [Article] [CrossRef] [PubMed] [Google Scholar]
- Li W, Wang Z, Deschler F, et al. Chemically diverse and multifunctional hybrid organic-inorganic perovskites. Nat Rev Mater 2017; 2: 16099. [Article] [Google Scholar]
- Cheng P, Wang Y, Wang P, et al. Internally and externally induced chiral hybrid metal halide materials for advanced chiroptoelectronic applications. Chem Soc Rev 2026; 55: 2024-2069. [Article] [Google Scholar]
- Yuan M, Quan LN, Comin R, et al. Perovskite energy funnels for efficient light-emitting diodes. Nat Nanotech 2016; 11: 872-877. [Article] [Google Scholar]
- Long G, Sabatini R, Saidaminov MI, et al. Chiral-perovskite optoelectronics. Nat Rev Mater 2020; 5: 423-439. [Article] [Google Scholar]
- Wang Y, Zhang Y, Zhang P, et al. High intrinsic carrier mobility and photon absorption in the perovskite CH3NH3PbI3. Phys Chem Chem Phys 2015; 17: 11516-11520. [Article] [Google Scholar]
- Sutherland BR, Sargent EH. Perovskite photonic sources. Nat Photon 2016; 10: 295-302. [Article] [Google Scholar]
- Stranks SD, Eperon GE, Grancini G, et al. Electron-hole diffusion lengths exceeding 1 micrometer in an organometal trihalide perovskite absorber. Science 2013; 342: 341-344. [Article] [CrossRef] [PubMed] [Google Scholar]
- Dong Q, Fang Y, Shao Y, et al. Electron-hole diffusion lengths > 175 μm in solution-grown CH3NH3PbI3 single crystals. Science 2015; 347: 967-970. [Article] [Google Scholar]
- Tian W, Zhao C, Leng J, et al. Visualizing carrier diffusion in individual single-crystal organolead halide perovskite nanowires and nanoplates. J Am Chem Soc 2015; 137: 12458-12461. [Article] [Google Scholar]
- Morad V, Stelmakh A, Svyrydenko M, et al. Designer phospholipid capping ligands for soft metal halide nanocrystals. Nature 2024; 626: 542-548. [Article] [Google Scholar]
- Bekenstein Y, Koscher BA, Eaton SW, et al. Highly luminescent colloidal nanoplates of perovskite cesium lead halide and their oriented assemblies. J Am Chem Soc 2015; 137: 16008-16011. [Article] [Google Scholar]
- Wang D, Li Y, Yang Y, et al. Energetic disorder dominates optical properties and recombination dynamics in tin-lead perovskite nanocrystals. eScience 2025; 5: 100279. [Article] [Google Scholar]
- Filip MR, Eperon GE, Snaith HJ, et al. Steric engineering of metal-halide perovskites with tunable optical band gaps. Nat Commun 2014; 5: 5757. [Article] [Google Scholar]
- Xu J, Boyd CC, Yu ZJ, et al. Triple-halide wide–band gap perovskites with suppressed phase segregation for efficient tandems. Science 2020; 367: 1097-1104. [Article] [Google Scholar]
- Prasanna R, Gold-Parker A, Leijtens T, et al. Band gap tuning via lattice contraction and octahedral tilting in perovskite materials for photovoltaics. J Am Chem Soc 2017; 139: 11117-11124. [Article] [Google Scholar]
- Lee MM, Teuscher J, Miyasaka T, et al. Efficient hybrid solar cells based on meso-superstructured organometal halide perovskites. Science 2012; 338: 643-647. [Article] [CrossRef] [PubMed] [Google Scholar]
- Jeon NJ, Noh JH, Kim YC, et al. Solvent engineering for high-performance inorganic-organic hybrid perovskite solar cells. Nat Mater 2014; 13: 897-903. [Article] [Google Scholar]
- Qin P, Paek S, Dar MI, et al. Perovskite solar cells with 12.8% efficiency by using conjugated quinolizino acridine based hole transporting material. J Am Chem Soc 2014; 136: 8516-8519. [Article] [Google Scholar]
- Jiang Y, Sun C, Xu J, et al. Synthesis-on-substrate of quantum dot solids. Nature 2022; 612: 679-684. [Article] [Google Scholar]
- Lin K, Xing J, Quan LN, et al. Perovskite light-emitting diodes with external quantum efficiency exceeding 20 per cent. Nature 2018; 562: 245-248. [Article] [Google Scholar]
- Quan LN, Yuan M, Comin R, et al. Ligand-stabilized reduced-dimensionality perovskites. J Am Chem Soc 2016; 138: 2649-2655. [Article] [Google Scholar]
- Saidaminov MI, Adinolfi V, Comin R, et al. Planar-integrated single-crystalline perovskite photodetectors. Nat Commun 2015; 6: 8724. [Article] [Google Scholar]
- Ma Y, Xu X, Li T, et al. Amplified narrowband perovskite photodetectors enabled by independent multiplication layers for anti-interference light detection. Sci Adv 2025; 11: eadq1127. [Article] [Google Scholar]
- Zou C, Ren Z, Hui K, et al. Electrically driven lasing from a dual-cavity perovskite device. Nature 2025; 645: 369-374. [Article] [Google Scholar]
- Veldhuis SA, Boix PP, Yantara N, et al. Perovskite materials for light-emitting diodes and lasers. Adv Mater 2016; 28: 6804-6834. [Article] [Google Scholar]
- Fu J, Bian T, Yin J, et al. Organic and inorganic sublattice coupling in two-dimensional lead halide perovskites. Nat Commun 2024; 15: 4562. [Article] [Google Scholar]
- Li Q, Zhang Y, Zhang G, et al. Recent advances in the development of perovskite@metal-organic frameworks composites. NSO 2023; 2: 20220065. [Article] [Google Scholar]
- Mihalyi-Koch W. Tuning the symmetry of hybrid organic-inorganic materials. Chem 2024; 10: 1965-1967. [Article] [Google Scholar]
- Dou Y, Wang X, Smith NWG, et al. Long-lived photoinduced polar states in metal halide perovskites. Nat Commun 2025; 16: 7230. [Article] [Google Scholar]
- Hautzinger MP, Mihalyi-Koch W, Jin S. A-site cation chemistry in halide perovskites. Chem Mater 2024; 36: 10408-10420. [Article] [Google Scholar]
- Zheng H, Loh KP. Ferroics in hybrid organic–inorganic perovskites: Fundamentals, design strategies, and implementation. Adv Mater 2024; 36: 2308051. [Article] [Google Scholar]
- Zhao BQ, Li Y, Chen XY, et al. Engineering carrier dynamics in halide perovskites by dynamical lattice distortion. Adv Sci 2023; 10: 2300386. [Article] [Google Scholar]
- Doherty TAS, Nagane S, Kubicki DJ, et al. Stabilized tilted-octahedra halide perovskites inhibit local formation of performance-limiting phases. Science 2021; 374: 1598-1605. [Article] [Google Scholar]
- Cheng J, Yi G, Zhang Z, et al. In situ chiral template approach to synthesize homochiral lead iodides for second-harmonic generation. Angew Chem Int Ed 2024; 63: e202318385. [Article] [Google Scholar]
- Schmitt T, Bourelle S, Tye N, et al. Control of crystal symmetry breaking with halogen-substituted benzylammonium in layered hybrid metal-halide perovskites. J Am Chem Soc 2020; 142: 5060-5067. [Article] [Google Scholar]
- Zhu C, Jin J, Wang Z, et al. Supramolecular assembly of blue and green halide perovskites with near-unity photoluminescence. Science 2024; 383: 86-93. [Article] [Google Scholar]
- Luo X, Liu W, Wang Z, et al. Thermally driven phase transition of halide perovskites revealed by big data-powered in situ electron microscopy. J Chem Phys 2023; 158: 134705. [Article] [Google Scholar]
- Leonard AA, Diroll BT, Flanders NC, et al. Light-induced transient lattice dynamics and metastable phase transition in CH3NH3PbI3 nanocrystals. ACS Nano 2023; 17: 5306-5315. [Article] [Google Scholar]
- Lang F, Pang J, Bu XH. Stimuli-responsive coordination polymers toward next-generation smart materials and devices. eScience 2024; 4: 100231. [Article] [Google Scholar]
- Wu J, Liu SC, Li Z, et al. Strain in perovskite solar cells: Origins, impacts and regulation. Natl Sci Rev 2021; 8: nwab047. [Article] [Google Scholar]
- Zhu H, Teale S, Lintangpradipto MN, et al. Long-term operating stability in perovskite photovoltaics. Nat Rev Mater 2023; 8: 569-586. [Article] [Google Scholar]
- Zheng N, Cao S, Zhang T, et al. Synthesis of zero-dimensional octahedral metal halides through solvent incorporation and their photophysical properties. Nat Chem 2025; 17: 1401-1409. [Article] [Google Scholar]
- Guo TM, Gao FF, Gong YJ, et al. Chiral two-dimensional hybrid organic–inorganic perovskites for piezoelectric ultrasound detection. J Am Chem Soc 2023; 145: 22475-22482. [Article] [Google Scholar]
- Han W, Cheng P, Guan J, et al. Extendable synthesis of organic cations for in situ construction of hybrid metal halides with near-unity photoluminescence and strong second harmonic generation. Angew Chem Int Ed 2025; 64: e202500786. [Article] [Google Scholar]
- Li XY, Wei Q, Hu CL, et al. Achieving large second harmonic generation effects via optimal planar alignment of triangular units. Adv Funct Mater 2023; 33: 2210718. [Article] [Google Scholar]
- Kim J, Kimura Y, Puchala B, et al. Dissolution enables dolomite crystal growth near ambient conditions. Science 2023; 382: 915-920. [Article] [Google Scholar]
- Hellmann R, Cotte S, Cadel E, et al. Nanometre-scale evidence for interfacial dissolution–reprecipitation control of silicate glass corrosion. Nat Mater 2015; 14: 307-311. [Article] [Google Scholar]
- Calligaris M, Faleschini P. Dimethylacetonylsulfonium tetrachlorobis(dimethyl sulfoxide-S)rhodate(III). Acta Crystlogr C Cryst Struct Commun 1995; 51: 588-591. [Article] [Google Scholar]
- Rudnitskaya OV, Tereshina TA, Dobrokhotova EV, et al. Chemical evolution in solutions of Ir complex [H(dmso)2]2[IrCl6]. Structures of [H(dmso)2]2[IrCl6], [H(dmso)][IrCl4(dmso)2], [Me2SCH2C(O)Me][IrCl4(dmso)2], [Me2SCH2C(O)-Me]2[IrCl6] and its Os analogue. Eur J Inorg Chem 2022; : e202200463. [Article] [Google Scholar]
- Zhu P, Wang D, Zhang Y, et al. Aqueous synthesis of perovskite precursors for highly efficient perovskite solar cells. Science 2024; 383: 524-531. [Article] [Google Scholar]
- Guan J, Zheng Y, Cheng P, et al. Free halogen substitution of chiral hybrid metal halides for activating the linear and nonlinear chiroptical properties. J Am Chem Soc 2023; 145: 26833-26842. [Article] [Google Scholar]
- Yao F, Peng J, Li R, et al. Room-temperature liquid diffused separation induced crystallization for high-quality perovskite single crystals. Nat Commun 2020; 11: 1194. [Article] [Google Scholar]
- Shi D, Adinolfi V, Comin R, et al. Low trap-state density and long carrier diffusion in organolead trihalide perovskite single crystals. Science 2015; 347: 519-522. [Article] [Google Scholar]
- Smith IWM. The temperature-dependence of elementary reaction rates: Beyond Arrhenius. Chem Soc Rev 2008; 37: 812-826. [Article] [Google Scholar]
- Kaiser D, Klose I, Oost R, et al. Bond-forming and -breaking reactions at sulfur(IV): Sulfoxides, sulfonium salts, sulfur ylides, and sulfinate salts. Chem Rev 2019; 119: 8701-8780. [Article] [Google Scholar]
- Agarwal T, Prasad AK, Advani SG, et al. Infrared spectroscopy for understanding the structure of Nafion and its associated properties. J Mater Chem A 2024; 12: 14229-14244. [Article] [Google Scholar]
- Hadjiivanov KI, Panayotov DA, Mihaylov MY, et al. Power of infrared and Raman spectroscopies to characterize metal-organic frameworks and investigate their interaction with guest molecules. Chem Rev 2021; 121: 1286-1424. [Article] [Google Scholar]
- Tauc J, Grigorovici R, Vancu A. Optical properties and electronic structure of amorphous germanium. Physica Status Solidi (B) 1966; 15: 627-637. [Article] [Google Scholar]
- Tauc J. Optical properties and electronic structure of amorphous Ge and Si. Mater Res Bull 1968; 3: 37-46. [Article] [Google Scholar]
- Saparov B, Mitzi DB. Organic–inorganic perovskites: Structural versatility for functional materials design. Chem Rev 2016; 116: 4558-4596. [Article] [Google Scholar]
- Hu YQ, Hui HY, Lin WQ, et al. Crystal and band-gap engineering of one-dimensional antimony/bismuth-based organic–inorganic hybrids. Inorg Chem 2019; 58: 16346-16353. [Article] [Google Scholar]
- Senocrate A, Kim GY, Grätzel M, et al. Thermochemical stability of hybrid halide perovskites. ACS Energy Lett 2019; 4: 2859-2870. [Article] [Google Scholar]
- Nowok A, Sobczak S, Roszak K, et al. Temperature and volumetric effects on structural and dielectric properties of hybrid perovskites. Nat Commun 2024; 15: 7571. [Article] [Google Scholar]
- Han X, Cheng P, Han W, et al. Circularly polarized luminescence and nonlinear optical harmonic generation based on chiral zinc halides. Chem Commun 2023; 59: 7447-7450. [Article] [Google Scholar]
- Morad V, Shynkarenko Y, Yakunin S, et al. Disphenoidal zero-dimensional lead, tin, and germanium halides: Highly emissive singlet and triplet self-trapped excitons and X-ray scintillation. J Am Chem Soc 2019; 141: 9764-9768. [Article] [Google Scholar]
- Chen Y, Zhou L, Zhou S, et al. Effect of the host lattice environment on the expression of 5s2 lone-pair electrons in a 0D bismuth-based metal halide. Inorg Chem 2023; 62: 2806-2816. [Article] [Google Scholar]
- Yuan C, Li X, Semin S, et al. Chiral lead halide perovskite nanowires for second-order nonlinear optics. Nano Lett 2018; 18: 5411-5417. [Article] [Google Scholar]
- Xiong J, Li X, Yuan C, et al. Wavelength dependent nonlinear optical response of tetraphenylethene aggregation-induced emission luminogens. Mater Chem Front 2018; 2: 2263-2271. [Article] [Google Scholar]
- Robinson K, Gibbs GV, Ribbe PH. Quadratic elongation: A quantitative measure of distortion in coordination polyhedra. Science 1971; 172: 567-570. [Article] [Google Scholar]
- Huang H, Yang Y, Qiao S, et al. Accommodative organoammonium cations in A-sites of Sb-In halide perovskite derivatives for tailoring broadband photoluminescence with X-ray scintillation and white-light emission. Adv Funct Mater 2024; 34: 2309112. [Article] [Google Scholar]
All Figures
|
Scheme 1 Schematic illustration of the correspondence between the sequential generation of intermediate and product cations from the DMSO and acetone condensation reaction and the sequential formation of the three corresponding OIHMH crystals. |
| In the text | |
|
Figure 1 Multistep structural evolution of OIHMHs driven by the in-situ condensation reaction. (a) Schematic illustration for the formation of various OIHMH crystal structures achieved by direct employment of the DMSO and acetone reaction pathway as the liquid diffusion crystallization system. (b) Structures of three distinct types of obtained OIHMHs, crystallized in the C2/c, Pnma and R-3m space groups, respectively. (c) Schematic illustration for the purification of the three OIHMH crystal types achieved by designing the DMSO and acetone reaction system as a vapor diffusion crystallization. (d) The sequential incorporation of the intermediate and product cations into the A-sites constructs three distinct OIHMH structures. (e) The minimum asymmetric units and structural transformation sequence of the three obtained OIHMH types. H atoms are omitted for clarity. |
| In the text | |
|
Figure 2 Spectroscopic characterizations. (a) 1H NMR spectra of Sb-based OIHMH I, OIHMH II and OIHMH III, in DMSO-d6. (b) 13C NMR spectra of the Bi/Sb-based OIHMH II dissolved samples, in DMSO-d6. (c) FTIR spectra of the three Sb-based OIHMH powders. (d) UV-vis transmission spectra and Eg comparison of the three types of Bi/Sb-based OIHMHs. (e) Comparison of the thermal decomposition temperature of the three types of Bi/Sb-based OIHMHs. |
| In the text | |
|
Figure 3 Variation of optical properties during the crystal transition. (a) Normalized PL spectra of the three Sb-based OIHMH crystals. (b) Comparison of the PL decays and PLQYs. (c) Comparison of TPF, at their optimal incident laser wavelength, with a laser power of 50 mW. (d) Crystal transition of the Sb-based OIHMH monitored through UV-light crystal images and corresponding PXRD patterns. |
| In the text | |
|
Figure 4 Structural comparison of the inorganic components among the three types of OIHMHs. The metal–metal distances between adjacent inorganic components of Sb-based OIHMH I (a), OIHMH II (b) and OIHMH III (c). The bond lengths, bond angles, and distortions of the [SbBr6]3− and [BiBr6]3− inorganic components across OIHMH I (d), OIHMH II (e), and OIHMH III (f). |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.